Amazon for the Integrative Genomics Viewer (IGV)
TL;DR: https://github.com/igvteam/igv/pull/620
Intro
In bioinformatics there’s hardly anybody that hasn’t come across the Broad Institute’s venerable Java Integrative Genomics Viewer, if not, have a peek at the original nature methods publication: it wouldn’t be an overstatement to say that IGV is the samtools of genome visualization.
Even Google’s deepvariant used IGV to validate their algorithms and learn about variants:
“The read and reference data are encoded as an image for each candidate variant site (…) These labeled image + genotype pairs, along with an initial CNN, which can be a random model, a CNN trained for other image classification tests (…) the reference and read bases, quality scores, and other read features are encoded into a red–green–blue (RGB) pileup image at a candidate variant”
Originally conceived as a locally-run desktop Java application IGV is, still today, the bread and butter of bioinformaticians and clinicians alike. With the arrival of whole genome sequencing (WGS), datasets became bigger so time consuming storage logistics burdens started to creep up.
Analyzing several WGS datasets quickly turned into a bioinformatics sweat shop pattern. To solve that, IGV learned new tricks to access remote data, but how good those turned out to be in the research trenches?
The typical IGV setup
The implementation of such tricks hacks require a backend with a traditional client/server model. The server is talking to IGV via HTTP (range) requests and requesting only a handful of genomic reads that the user needs to see at a particular genomic region. Since 24/7 longrunning processes often clash with the traditional batch-oriented HPC culture, research software engineers have resorted to suboptimal workarounds to expose the valued sequencing data to their colleagues:
- SSH+X11 forwarding: Low resolution and overall sluggish UI.
- SSHFS-related user level mounts: Prone to filesystem stalling when your laptop briefly goes to sleep.
- S3 FUSE mount on AWS via S3Proxy or similar: Majorly unused EC2 instance, low network throughput if that instance is not on the higher end (wasteful, expensive proposition).
Worth mentioning, the genomic viewer alternatives, NGB, jbrowse, IGB and IGV.js unfortunately have some pain points:
- They currently implement a traditional client-server model which leads to the aforementioned waste of (backend) resources.
- Some are under-mantained (funding got cut?).
- Their feature set is “not there yet”, or there’s “that” missing feature that conservative clinicians trust and love.
- Their software architecture has questionable choices that endanger its future sustainability.
- All of the above or worse.
So what’s left when, as Titus Brown puts it:
“Scientific culture is generational; change will come, but slowly”
There must be a better way for users to overcome those data access barriers while lowering CO2 emissions and keeping everything safe, right?
Our answer is to adapt what’s most used (IGV desktop) to a more modern and efficient paradigm, hopefully transitioning to a better situation in the future, incrementally.
So we connected IGV desktop directly to our our main cloud provider. At UMCCR that is Amazon’s AWS.
Next section for the tech gory details if you are still here. Jump towards the end for new possibilities to awe for :)
Getting genomic reads directly from S3 into IGV
Right after forking IGV from upstream into an aws_support branch, we tackled the backend, navigating the methods with IDEA IntelliJ community edition, unfortunately VSCode is quite far behind with Java editor ergonomy.
Backend
All our cancer sequencing data is deposited in S3 buckets with encryption at rest. Furthermore, in order to access the data, researchers have to authenticate and get authorized via AWS Cognito, which supports federated authentication via Google in our particular case but others are also supported out of the box. Our IGV auth flow looks like this (see images in tweet):
I implemented @awscloud S3 support (plus federated auth via #AWS #cognito) into the @igvteam Java desktop client. Some rough edges left to fix, but it works quite well, check it out on the ongoing PR: https://t.co/HRzIEVTfxH /cc @delagoya @fiamh @ga4gh @umccr pic.twitter.com/Aps1BSI3nJ
— a.k.a brainstorm (@braincode) January 30, 2019
Under the hood, and after the OAuth/OIDC flow, a temporary AWS STS token is issued to give IGV access to a particular AWS resource:
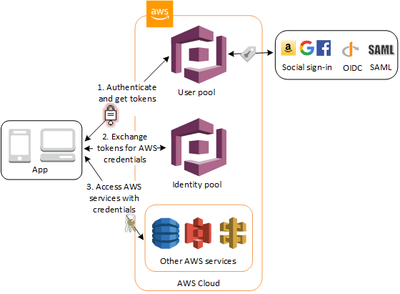
A policy to limit access to a particular bucket is defined in the AWS cognito “authenticated users” default IAM role:
{
"Version": "2012-10-17",
"Statement": [
{
"Sid": "IGVCognitoAuthedUsers",
"Effect": "Allow",
"Action": [
"s3:ListBucket",
"s3:GetObject"
],
"Resource": [
"arn:aws:s3:::your_bucket",
"arn:aws:s3:::your_bucket/*"
]
},
{
"Sid": "IGVListBuckets",
"Effect": "Allow",
"Action": [
"s3:ListAllMyBuckets"
],
"Resource": [
"arn:aws:s3:::*"
]
}
]
}
The default unauthed cognito role is left empty so those users do not get access to anything and since IAM roles and policies have a default “deny all” policy.
Using the AWS SDK for Java we abstracted away the common misconception that AWS S3 is “like a filesystem”. It is not, it’s an object store, therefore this bit requires some attention so that the UI (and users) can deal with it:
The Amazon S3 data model is a flat structure: you create a bucket, and the bucket stores objects. There is no hierarchy of subbuckets or subfolders; however, you can infer logical hierarchy using key name prefixes and delimiters as the Amazon S3 console does. The Amazon S3 console supports a concept of folders.
You can browse most of this implementation in the AmazonUtils.java class on the ongoing IGV pullrequest.
UI
Since our growing data warehouse has many objects, we were concerned that retrieving a s3 bucket tree listing recursively at once could become too slow from an UX/UI perspective. Therefore, we implemented JTree’s TreeWillExpand listener so that S3 bucket listings could be obtained dynamically as the user explored the potentially many levels of the hierarchy, wasting no API calls nor time.
The current hardcoded/mock/prototype GA4GH IGV data model and corresponding UI representation gave us an idea of how to shoehorn our dynamic approach.
About feeding giants and possible bright(er) futures
Now, we hear you say: why are we feeding Mr Bezos giant machine when we have local HPC produce?
Well, in some situations, uptime is not counted in 9’s but perhaps in 8’s or 0’s if you are lucky… true story:
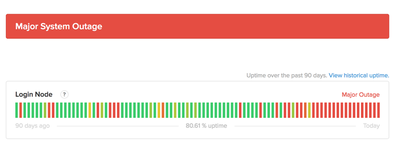
We would like to think that bioinformatics is transitioning towards consolidation, managed infrastructure that we can rely on and paying the commercial cloud premium is a good way towards that reality. We would like to go forward where fighting ancient, barely funded tools and infrastructure are not the norm anymore, but instead modern, integrated and robust workflows that gather metrics which allow increasing performance enable or staff to aim higher in their science. As an end result, tightening the loop between research question and results, iterating faster and overall having a better time as a researcher.
This particular serverless solution will probably be forgotten as newer developments arise, but at least it allowed us to very easily profile data access patterns and begin to decide which data to store on cheaper archival solutions (such as AWS Glacier), as opposed to rule of thumb, arbitrarily decided archival lifecycles. After this development we are beginning to switch from “where to connect”, “which data to host where” type of questions which plague core sequencing facilities to more interesting ones:
- Which genomic curators in our team make queries in our custom cancer gene list against which samples? Can we dynamically (re)generate that list based on IGV curator queries?
- How many and which regions/variants/SVs/SNPs are examined and how much time is spent on them per curator? Human fatigue can be a bias in cancer interpretation work, can we build better tools to avoid this situation?
- Shall we integrate variant sharing tools like VIPER to have robust quorum on cancer interpretation work?
We know that the Broad institute had significant pushback in the past when implementing its controversial metric-gathering GATK phone home feature, and let us be clear that none of those tracking/metric changes are present nor will be included in the current pullrequest against IGV proper but having this system in place has already helped us make informed decisions on our core activity in many levels in the few days it has been in production.
Next possible stop to insure better interoperability: htsget. Any takers?